







Population Genetics, Genomics and Metagenomic Analyses
Studying genetic differences of populations, genomes from communities and surveying microbial community using 16S rRNA gene studies.
Genomic Analyses to Understand the Diversity, Ecology and Evolution of Multipartite Microbial Symbioses
Jonathan Klassen studies microbial community ecology, especially using the fungus-growing ant symbiosis as a model system to understand how microbial interaction networks evolve. He is particularly interested in using relatively simple multipartite symbioses as models to understand the evolutionary and ecological rules governing the stability and function of microbial communities more generally. The Klassen lab uses culture-dependent and -independent genomics coupled with comparative phenotyping and chemical biology to understand how the differing ecologies and population structures of microbes govern the stability and function of the communities to which they belong.
Genomics, proteomics and transcriptomics of bacteria (actinomycetes) that infect higher plants and fix atmospheric nitrogen; diversity of fungi associated with surface ripened cheese
Benson's research interests include microbial biogeography, including the molecular and genomic interactions of bacteria with their environment, and the genomic evolution controlling distribution of microbes in several environments. The specific areas of interest include plant-microbe and insect-microbe microbiomes, and the distribution and diversity of microorganisms associated with cheese.
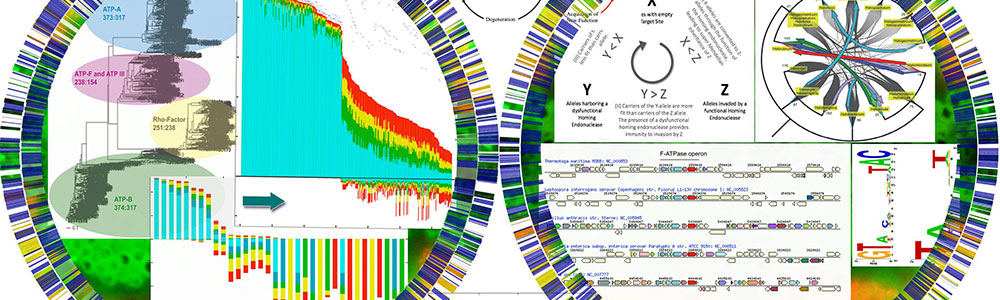
Molecular parasites (inteins and homing endonucleases) in microbial populations using microcosm and metagenomic approaches.
J. Peter Gogarten's lab studies the evolutionary histories of genes and organisms. Currently, the main research focus is comparative genomics and horizontal gene transfer. He also studies the early evolution of life (early expansion of the genetic code, gene family expansions, traces of extinction events in the molecular record) and the evolution of molecular parasites (inteins and homing endonucleases).
Population genealogical history (especially the effect the meiotic recombination) & Problems related to high-throughput sequencing
Yufeng Wu's research interests are algorithmic approaches in bioinformatics. His current research focus is on the development of new methods for population genomics and evolution, and high-throughput sequencing.
Understanding how genetic variation is distributed within and between haloarchaeal species and how molecular mechanisms for gene transfer work in Archaea
Papke lab Although we recognize names of microorganisms like Escherichia coli and Bacillus subtilis, our ability to classify strains into "natural kinds" is rigorously tested by the observation that genetic variation is frequently shuttled across so-called species boundaries. Also, much of what we know about species comes from well-studied pathogenic bacteria, which are often classified by the disease that they cause (e.g., Bacillus anthracis or Neisseria gonorrhoeae). This is important to doctors and their patients but the Earth¹s biomass and diversity is comprised mainly of prokaryotes, the vast majority of which do not cause disease. Combined, these observations suggest that our understanding of species evolution is relatively shallow and that our current standards of classification are biased and difficult to apply to most microbes. The main goal of our research is to use evolution and ecology theory in combination with approaches like genomics, metagenomics and population genetics to investigate intra and inter species variation, gene flow and genetic relationships for non-pathogenic (e.g., environmental) prokaryotes. We concentrate on hypersaline adapted Archea (Haloarchaea) as model organisms because they live in island-like habits which aid in simplifying or sorting the evolutionary forces that effect their distributions, adaptations and variation.